El síndrome de Papillon-Lefèvre (PLS, por sus siglas en inglés) es un trastorno genético extremadamente raro que se manifiesta típicamente a partir de aproximadamente uno a cinco años de edad. El PLS se caracteriza por el desarrollo de manchas escamosas y secas en la piel de las palmas de las manos y las plantas de los pies (hiperqueratosis palmar-plantar) en asociación con inflamación severa y degeneración de las estructuras que rodean y sostienen los dientes (periodonto).
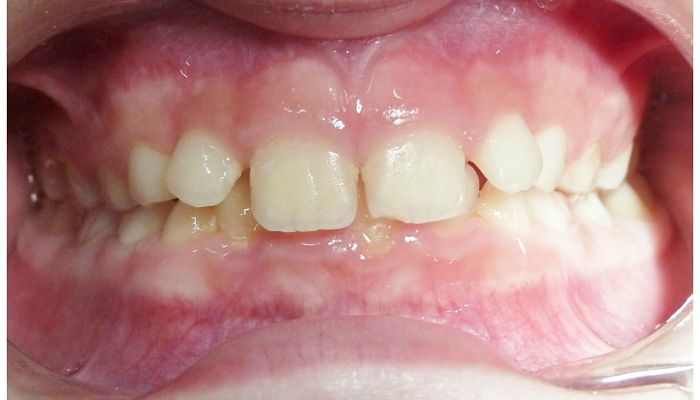
Los dientes primarios (caducos) con frecuencia se aflojan y se caen hacia la edad de cinco años. Sin tratamiento, la mayoría de los dientes secundarios (permanentes) también pueden perderse aproximadamente a los 17 años. Los síntomas y hallazgos adicionales asociados con el PLS pueden incluir infecciones frecuentes de la piel que producen pus (piogénicas), anormalidades de las uñas (distrofia de las uñas) y sudoración excesiva (hiperhidrosis).
El síndrome de Papillon-Lefèvre se transmite como un rasgo autosómico recesivo. Es el resultado de cambios (alteraciones) del gen CTSC que regula la producción de una enzima conocida como catepsina C.
Signos y síntomas
El síndrome de Papillon-Lefèvre se caracteriza por el desarrollo de manchas de piel secas y escamosas (hiperqueratosis) generalmente alrededor de la edad de uno a cinco años.
Estos parches generalmente se limitan a la parte inferior de las manos y los pies, pero pueden extenderse a las rodillas y los codos. En algunas personas raras, las partes superiores de las manos y los pies, los párpados, los labios, las mejillas y/u otras áreas del cuerpo también pueden verse afectadas.
La piel afectada puede estar inusualmente roja y gruesa, pero puede variar en color y textura. Las lesiones cutáneas pueden empeorar con el frío y caminar puede ser doloroso. En raras ocasiones, se pueden formar quistes en los párpados posteriormente en la vida.
En las personas afectados, los dientes generalmente parecen formarse y erupcionar normalmente. Sin embargo, la mayoría de las personas afectadas presentan inflamación crónica severa y degeneración de los tejidos que rodean y sostienen los dientes (gingivitis y periodontitis).
Las encías y los ligamentos y huesos subyacentes que soportan los dientes generalmente están comprometidos. Cuando los dientes de leche (caducos o primarios) erupcionan, estas áreas se enrojecen, se hinchan y sangran (gingivitis).
La boca puede inflamarse (estomatitis), los ganglios linfáticos pueden hincharse (adenopatía regional) y se pueden formar bolsas en las encías que causan susceptibilidad a las infecciones. Se puede desarrollar un mal aliento notable, llamado halitosis.
Masticar puede ser muy doloroso. La cresta ósea engrosada en la que descansan los dientes (proceso alveolar) puede romperse (lisis ósea alveolar). Los dientes de leche con frecuencia se aflojan y se caen a la edad de cuatro o cinco años.
Después, la inflamación puede reducirse y las encías pueden parecer más sanas. Sin embargo, sin tratamiento, la mayoría de los dientes adultos (permanentes) pueden perderse de la misma manera a la edad de 16 años.
Tanto los dientes de leche como los de adulto suelen verse afectados en el orden de su erupción. Algunos individuos afectados sólo tendrán enfermedad periodontal leve, y/o tendrán un inicio posterior de la enfermedad periodontal.
Los pacientes pueden tener infecciones cutáneas (piogénicas) que se repiten con frecuencia. Algunos individuos pueden estar en riesgo de otras infecciones incluyendo abscesos hepáticos, infección de los folículos pilosos (furunculosis), infecciones del tracto respiratorio e hidradenitis supportiva, una condición caracterizada por lesiones inflamadas y dolorosas que ocurren en la axila, la ingle, el ano y las regiones de los senos.
La mayoría de las personas tienen uñas frágiles que pueden romperse fácilmente. El vello en el cuero cabelludo y en el cuerpo puede ser escaso (hipotricosis). Algunos individuos pueden desarrollar cantidades excesivas de queratina en los folículos pilosos causando protuberancias (pápulas) ásperas en forma de cono en la piel. Las personas afectadas también pueden presentar sudoración excesiva (hiperhidrosis) asociada con un olor desagradable (maloliente). Algunos individuos experimentan la acumulación de calcio con el cráneo haciendo que el tejido afectado se endurezca (calcificación intracraneal).
Causas del síndrome de Papillon-Lefèvre
El síndrome de Papillon-Lefèvre es causado por cambios (alteraciones) en el gen de la catepsina C o CTSC. Los genes proporcionan instrucciones para crear proteínas que juegan un papel crítico en muchas funciones del cuerpo. Cuando se altera un gen, el producto proteico puede ser defectuoso, ineficiente o estar ausente. Dependiendo de las funciones de la proteína en particular, esto puede afectar a muchos sistemas de órganos del cuerpo.
El gen CTSC regula (codifica para) la producción de un tipo específico de enzima (proteasa lisosómica) conocida como catepsina C. La proteína se expresa a altos niveles en varias células inmunitarias y ciertas áreas corporales afectadas por el PLS.
Éstas incluyen las células fuertemente comprimidas, conocidas como células epiteliales, que forman la capa protectora externa de la piel (epidermis), como las palmas de las manos, las plantas de los pies y las rodillas, así como ciertas células de las encías (gingiva).
Se han detectado varias alteraciones diferentes del gen CTSC en las especies afectadas. Ciertas alteraciones pueden resultar en la pérdida casi completa de la actividad enzimática de la catepsina C en individuos con la enfermedad, o una actividad relativamente reducida de la enzima en algunos miembros de la familia que portan una sola copia alterada del gen (portadores heterocigotos).
Los investigadores creen que otros factores además de las alteraciones en el gen CTSC son necesarios para el desarrollo del síndrome de Papillon-Lefèvre. Lo más probable es que haya defectos en el sistema inmunológico, específicamente en la sangre blanca llamada neutrófilos o células asesinas naturales.
Los neutrófilos juegan un papel importante en ayudar al cuerpo a combatir las infecciones, especialmente las infecciones bacterianas y fúngicas. Las células asesinas naturales ayudan a combatir los virus. Sin embargo, la investigación de tales factores no ha llevado a hallazgos consistentes y se necesita más investigación para entender los mecanismos subyacentes que conducen al desarrollo del síndrome de Papillon-Lefèvre.
Síndrome de Papillon-Lefèvre heredado de forma autosómica recesiva. La mayoría de las enfermedades genéticas se determinan por el estado de las dos copias de un gen, una recibida del padre y otra de la madre. Los trastornos genéticos recesivos ocurren cuando un individuo hereda dos copias de un gen anormal para el mismo rasgo, una de cada padre.
Si un individuo hereda un gen normal y un gen para la enfermedad, la persona será portadora de la enfermedad, pero generalmente no mostrará síntomas. El riesgo de que dos padres portadores transmitan el gen alterado y tengan un hijo afectado es del 25% en cada embarazo.
El riesgo de tener un hijo portador como los padres es del 50% con cada embarazo. La probabilidad de que un niño reciba genes normales de ambos padres es del 25%. El riesgo es el mismo para hombres y mujeres.
Los padres de varios individuos con el síndrome de Papillon-Lefèvre han estado estrechamente relacionados por la sangre (consanguínea). Todos los individuos portan 4-5 genes anormales.
Los padres que son parientes cercanos (consanguíneos) tienen una mayor probabilidad que los padres sin parentesco de tener el mismo gen anormal, lo cual aumenta el riesgo de tener hijos con un trastorno genético recesivo.
Poblaciones afectadas
El síndrome de Papillon-Lefèvre es un trastorno extremadamente raro que afecta a hombres y mujeres en igual número y se encuentra en todos los grupos étnicos. Se han reportado más de 200 casos en la literatura médica. En la población general, el trastorno ocurre en aproximadamente uno a cuatro individuos por millón.
Sin embargo, determinar la incidencia o prevalencia de un trastorno raro es extremadamente difícil. Las anomalías cutáneas asociadas con este trastorno pueden estar presentes al nacer (congénitas) o a la edad de cinco años. Otros síntomas suelen aparecer entre el tercer y quinto año de vida.
Trastornos Relacionados
Los síntomas de los siguientes trastornos pueden ser similares a los del síndrome de Papillon-Lefèvre. Las comparaciones pueden ser útiles para un diagnóstico diferencial:
El síndrome de Haim-Munk, también conocido como queratosis palmoplantaris con periodontopatía y onicogryphosis, es un trastorno hereditario poco frecuente. Se caracteriza por enrojecimiento y crecimiento excesivo de la piel de las palmas de las manos y las plantas de los pies (hiperqueratosis palmoplantar) y crecimiento excesivo de las uñas de las manos y los pies (onicogryphosis).
Las personas afectadas también pueden tener pies planos (pes planus); dedos de las manos y de los pies anormalmente largos y delgados (aracnodactilia); y/o una mayor sensibilidad a las temperaturas frías y a la pérdida de tejido óseo en los dedos de las manos y de los pies (acroesteólisis).
También puede haber degeneración de las estructuras que rodean y soportan los dientes. El síndrome de Haim-Munk se hereda como un rasgo genético autosómico recesivo. Algunos investigadores creen que el trastorno puede ser una variante del síndrome de Papillon-Lefèvre. (Para más información, elija “Haim Munk” como término de búsqueda en la Base de Datos de Enfermedades Raras.
Es un raro trastorno hereditario caracterizado por el desarrollo de piel seca y escamosa en las palmas de las manos y las plantas de los pies (queratosis palmoplantar), uñas frágiles y/o el desarrollo de quistes en los párpados.
Otros síntomas pueden incluir la pérdida temprana de los dientes primarios (caducos), la ausencia de algunos o todos los dientes permanentes (hipodoncia), y/o la falta de vello corporal y/o del cuero cabelludo (hipotricosis). Se cree que el síndrome de Schopf-Schulz-Passarge se hereda como un rasgo genético autosómico dominante.
La paquioniquia congénita (PC) es un raro trastorno cutáneo queratinizante heredado de manera autosómica dominante. Las características predominantes son dolor plantar severo, queratodermia palmoplantar (PPK) incluyendo callos con ampollas subyacentes y distrofia de uñas hipertrófica variable, a menudo acompañada de leuceratosis oral, quistes de varios tipos, hiperqueratosis folicular, hiperhidrosis palmoplantar y a veces dientes natales.
La PC es causada por una mutación en cualquiera de los cinco genes de queratina, KRT6A, KRT6B, KRT6C, KRT16 o KRT17. (Para más información, elija “Pachyonychia congénita” como término de búsqueda en la Base de Datos de Enfermedades Raras.
Diagnóstico
El diagnóstico del síndrome de Papillon-Lefèvre puede confirmarse mediante una evaluación clínica exhaustiva que incluye una historia clínica detallada del paciente y la identificación de los hallazgos físicos característicos. En algunos casos, las anormalidades de la piel pueden ser aparentes al nacer (congénitas) o durante la infancia, incluyendo anormalidades características de la piel en las palmas de las manos y las plantas de los pies.
En la mayoría de los casos, el diagnóstico del trastorno puede no confirmarse hasta que la inflamación y degeneración de los tejidos que rodean y sostienen los dientes (periodonto) sea evidente.
Esto generalmente ocurre entre el tercer y quinto año de vida, cuando los dientes de leche (caducos) comienzan a erupcionar. En muchas personas, las anomalías de la piel ocurren simultáneamente con la pérdida de los dientes. Además, la identificación de la acumulación anormal de calcio en el cráneo (calcificación intracraneal) puede ayudar a confirmar el diagnóstico.
El diagnóstico se puede hacer con un simple análisis de la orina de un bebé o de un niño (análisis urinario). La orina de un niño del que se sospecha que tiene el síndrome de Papillon-Lefèvre se analiza para ver si hay alguna actividad de la enzima catepsina C. El diagnóstico del trastorno se realiza con poca o ninguna actividad de esta enzima. Esto es extremadamente importante porque el diagnóstico temprano y el tratamiento oportuno pueden prevenir potencialmente la periodontitis agresiva, la pérdida de dientes y mejorar la calidad de vida general de las personas con el síndrome de Papillon-Lefèvre.
Las pruebas de genética molecular pueden confirmar el diagnóstico. Las pruebas de genética molecular pueden detectar alteraciones en el gen CTSC que se sabe que causa el síndrome de Papillon-Lefèvre, pero sólo están disponibles como servicio de diagnóstico en laboratorios especializados.
Terapias Estándar
El tratamiento está dirigido hacia los síntomas específicos que son aparentes en cada individuo. El tratamiento puede requerir los esfuerzos coordinados de un equipo de especialistas.
Los pediatras, cirujanos, médicos que evalúan y tratan problemas de la piel (dermatólogos), dentistas, equipo quirúrgico dental, que incluye un dentista pediátrico, un especialista en el tratamiento de trastornos que afectan el área que sostiene y rodea los dientes (periodoncista), y un especialista en la restauración y reemplazo de dientes (prostodoncista), y otros profesionales de la salud pueden necesitar planear sistemática y exhaustivamente el tratamiento de un niño afectado.
La asesoría genética puede ser beneficiosa para las personas afectadas y sus familias. También se recomienda el apoyo psicosocial para toda la familia.
Los médicos pueden vigilar cuidadosamente a las personas afectadas para ayudar a prevenir y asegurar la identificación temprana de la infección. En caso de infección de las encías, se puede prescribir terapia antibiótica. Se recomienda una higiene bucal adecuada y el uso de enjuagues bucales. Si los dientes se pierden, pueden ser reemplazados con implantes dentales.
Se ha encontrado un éxito limitado en el tratamiento de las anomalías cutáneas asociadas con lubricantes tópicos y humectantes (emolientes). Algunas veces, también se pueden utilizar medicamentos que descomponen la capa externa de la piel (queratolíticos) o esteroides tópicos, que reducen la inflamación de la piel. En algunos casos, la cirugía y los injertos de piel se pueden utilizar para aliviar los problemas de piel. El uso de antitranspirantes y desodorantes puede ayudar con la transpiración excesiva (hiperhidrosis).
Terapias de investigación
El tratamiento con vitamina A (retinoides) ha resultado útil para tratar a algunas personas con el síndrome de Papillon-Lefèvre. El etretinato, la isotretinoína y la acitretina se han utilizado para reducir la inflamación crónica de las encías, lo que resulta en una disminución de la pérdida de dientes en algunos individuos afectados.
En algunos casos, la vitamina A también puede aliviar las anormalidades de la piel y ayudar a minimizar las infecciones cutáneas recurrentes que producen pus (piogénicas) asociadas con el trastorno. La identificación temprana del síndrome de Papillon-Lefèvre es esencial para un tratamiento potencialmente efectivo con vitamina.
A (retinoides). Se deben realizar más investigaciones antes de poder determinar la seguridad y efectividad a largo plazo del uso de retinoides para tratar el síndrome de Papillon-Lefèvre.
También te puede interesar leer: Pie De Atleta: Causas, Diagnóstico, Síntomas, Tratamiento

Dermatologa. Especialista en enfermedades de la piel, pelo y uñas.