La enfermedad de Refsum, clásica o adulta, heredopatía atactica polyneuritiformis, deficiencia de ácido fitánico oxidasa y enfermedad de almacenamiento de ácido fitánico, es una enfermedad neurológica autosómica recesiva que resulta en la sobre acumulación de ácido fitánico en células y tejidos. Es uno de los varios trastornos que lleva el nombre del neurólogo noruego Sigvald Bernhard Refsum (1907-1991).
La enfermedad de Refsum es típicamente de inicio en la adolescencia y se diagnostica por niveles de ácido fitánico superiores al promedio. Los seres humanos obtienen el ácido fitánico necesario principalmente a través de la dieta. Todavía no está claro qué función desempeña el ácido fitánico fisiológicamente en los seres humanos, pero se ha descubierto que regula el metabolismo de los ácidos grasos en el hígado de los ratones.

Fisiopatología
La enfermedad de Refsum es un trastorno recesivo caracterizado por la oxidación alfa peroxisomal defectuosa del ácido fitánico. Por consiguiente, este ácido graso exógeno de cadena ramificada C20 (ácido 3, 7, 11,15-tetrametilhexadecanoico) se acumula en la sangre y los tejidos.
Es casi exclusivamente de origen exógeno y se obtiene principalmente de la clorofila vegetal alimentaria y, en menor medida, de fuentes animales. Los niveles de ácido fitánico en la sangre aumentan en pacientes con la enfermedad de Refsum.
Estos niveles son de 10-50 mg/dL, mientras que los valores normales son menores o iguales a 0.2 mg/dL, y representan del 5-30% de los lípidos séricos. La neurodegeneración mediada por ácidos grasos merece un examen más profundo.
El ácido fitánico sustituye a otros ácidos grasos, incluidos los esenciales como el ácido linoleico y el ácido araquidónico, en las moieties lipídicas de varios tejidos.
Esta situación conduce a una deficiencia de ácidos grasos esenciales, que se asocia con el desarrollo de ictiosis. Se ha localizado un gen de la enfermedad de Refsum, la fitanoilasa-CoA hidroxilasa (PHYH), en la banda 10p13 entre los marcadores D10S226 y D10S223.
La enfermedad de Refsum es genéticamente heterogénea, y hasta un 55% de los casos no están relacionados con el locus del gen PAHX en D10S547 a D10S223. Se ha descubierto que algunos pacientes presentan un defecto en la perforina (defecto PEX7).
Basado en lo anterior, se propuso que la enfermedad de Refsum en adultos podría ser dividida en tipos 1 y 2, dependiendo de cuál gen es defectuoso. Por lo tanto, la enfermedad de Refsum, al igual que otras enfermedades peroxisomales, es un síndrome heterogéneo. Recientemente, un modelo de ratón para la enfermedad de Refsum (Phyh ratón knockout por disrupción dirigida del gen PHYH). En humanos, el gen PHYH es de aproximadamente 21 kb y consiste en 9 exones y 8 intrones. Codifica una proteína de 38.6 kd.
Una forma infantil de la enfermedad de Refsum también existe y es un trastorno autosómico recesivo de la biogénesis peroxisomal, que conduce a muchas anomalías bioquímicas, incluyendo:
- Una elevada concentración plasmática de ácido fitánico, ácido pristánico, ácidos grasos de cadena muy larga y ácidos biliares C27.
- La enfermedad se presenta en el primer año de vida y se manifiesta con retraso en el desarrollo, trastornos visuales y auditivos y rasgos dismórficos.
- La ictiosis es un síntoma inusual.
Etiología
La enfermedad de Refsum es causada por mutaciones en los genes phytanoyl-CoA hidroxylase (PHYH) y el receptor PTS2 (PEX7). Este trastorno se hereda en forma autosómica recesiva. Un solo defecto de la enzima peroxisomal que causa deficiencia de alfa-oxidación conduce a la acumulación de ácido fitánico en la sangre y tejidos de pacientes con la enfermedad de Refsum. El efecto citotóxico del ácido fitánico parece deberse a una acción combinada de regulación de Ca2+, despolarización mitocondrial y aumento de la generación de especies reactivas de oxígeno en las células cerebrales.
También puedes leer: Schwannoma: Causas, Síntomas, Herencia,Tratamientos
Antecedentes
La enfermedad de Refsum (ER) es un síndrome neurocutáneo que se caracteriza bioquímicamente por la acumulación de ácido fitánico en plasma y tejidos. Los pacientes con la enfermedad de Refsum son incapaces de degradar el ácido fitánico debido a una actividad deficiente de la fitanoilasa-CoA hidroxilasa (PhyH), una enzima peroxisomal que cataliza el primer paso de la alfa-oxidación del ácido fitánico.
La enfermedad de Refsum puede clasificarse como un trastorno de biogénesis peroxisómica. Esta categoría se hereda como un rasgo autosómico recesivo y se caracteriza por la alteración del ensamblaje del peroxisoma, lo que resulta en múltiples deficiencias de la enzima peroxisoma, secuelas complejas del desarrollo y discapacidades progresivas. La enfermedad de Refsum infantil es un trastorno de biogénesis peroxisómica.
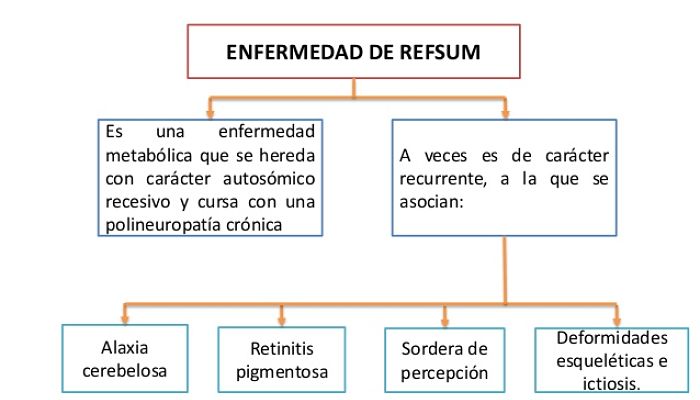
Refsum describió por primera vez esta enfermedad en 1946. La polineuropatía periférica, la ataxia cerebelosa, la retinitis pigmentaria y la ictiosis son los principales componentes clínicos. Los síntomas evolucionan lenta e insidiosamente desde la infancia hasta la adolescencia y la adultez temprana.
Características de la enfermedad de Refsum

Individuos con enfermedad de Refsum presentes con daño neurológico, degeneración cerebelosa y neuropatía periférica. El inicio es más común en la niñez/adolescencia con un curso progresivo, aunque ocurren períodos de estancamiento o remisión.
Los síntomas también incluyen ataxia, piel escamosa (ictiosis), dificultad para oír y problemas oculares como retinitis pigmentaria, cataratas y ceguera nocturna. En el 80% de los pacientes diagnosticados con la enfermedad de Refsum se ha reportado pérdida auditiva neurosensorial, es decir, pérdida auditiva como resultado de daño al oído interno o al nervio conectado al oído con el cerebro.
Causa
La enfermedad de Refsum es un trastorno peroxisomal causado por la alteración de la oxidación alfa de los ácidos grasos de cadena ramificada, que provoca la acumulación de ácido fitánico y sus derivados en el plasma y los tejidos.
Esto puede deberse a deficiencias de fitanoil-CoA hidroxilasa o actividad de la peroxina-7. En general, la enfermedad de Refsum es causada por mutaciones de PHYH. Las mutaciones del gen PEX7 pueden interrumpir el transporte peroxisómico de proteínas, ya que este gen codifica el receptor de la proteína peroxina 7.
Estas mutaciones en el gen PEX7 generalmente conducen a la condrodisplasia punctata rizomélica tipo 1- que afecta el desarrollo de muchas partes del cuerpo. La enfermedad de Refsum se hereda en un patrón autosómico recesivo, lo que significa que requiere ambas copias de la mutación para heredar la enfermedad.
Diagnóstico
Histopatología, la piel muestra hiperqueratosis, hipergranulosis y acantosis. Los hallazgos patognomónicos ocurren en las células basales y suprabasales de la epidermis, las cuales muestran vacuolas de tamaño variable que contienen acumulaciones de lípidos.
Frecuencia

La enfermedad de Refsum es rara, con sólo 60 casos publicados en todo el mundo.
- Carrera. No hay predominio racial.
- Sexo. Inicialmente sólo se reportaron casos de hombres; sin embargo, ahora no predomina ninguno de los dos sexos.
- Edad. La enfermedad clásica de Refsum se manifiesta en niños de 2 a 7 años de edad; sin embargo, el diagnóstico generalmente se retrasa hasta la edad adulta temprana. Sin embargo, se describió a un paciente con una rara enfermedad adulta de inicio tardío que se hizo evidente por primera vez a la edad de 72 años. La enfermedad de Refsum infantil aparece en la primera infancia.
Pronóstico
El pronóstico en pacientes no tratados generalmente es deficiente. La disfunción de las fibras nerviosas mielinizadas y del sistema de conducción cardíaca conduce a síntomas neuropáticos centrales y periféricos, deterioro de la visión y arritmias cardíacas. Estos últimos son con frecuencia la causa de la muerte. Se ha descrito neuropatía vestibular reversible.
En los casos diagnosticados y tratados tempranamente, el ácido fitánico disminuye lentamente, seguido de una mejora de la descamación de la piel y, en un grado variable, de la reversión de los signos neurológicos recientes. Se informa sobre la retención de la visión y la audición.
La atenuación de los síntomas neurológicos, oftalmológicos y cardíacos requiere una adhesión constante a una dieta adecuada y plasmaféresis.
Clasificación
- La enfermedad de Refsum del adulto puede dividirse en los subtipos de enfermedad de Refsum del adulto 1 y enfermedad de Refsum del adulto 2.
- La primera proviene de mutaciones en el gen phytanoyl-CoA hydroxylase (PAHX aka PHYH), en el locus PHYH a 10p13 en el cromosoma 6q22-24.
- Inicialmente se creía que esta era la única mutación; sin embargo, el 55% de los casos se atribuyen ahora a mutaciones en otros genes.
- La enfermedad de Refsum proviene de mutaciones en el gen de la peroxina.
- Esta mutación en el gen PEX7 también se encuentra en el cromosoma 6q22-24, y se encontró en pacientes que presentaban acumulación de ácido fitánico sin mutación de PHYH.
La enfermedad de Refsum adulta no debe confundirse con la enfermedad de Refsum infantil, un trastorno de biogénesis peroxisómica resultante de deficiencias en el catabolismo de ácidos grasos de cadena muy larga y ácidos grasos de cadena ramificada (como el ácido fitánico) y la biosíntesis del plasmalógeno.
Tratamiento
Dado que el ácido fitánico no se produce de forma endógena en el cuerpo humano, los individuos con la enfermedad de Refsum suelen seguir una dieta restringida por el ácido fitánico y evitan el consumo de grasas de rumiantes y de ciertos peces, como el atún, el bacalao y el eglefino.
También se evitan los animales que se alimentan de pasto y su leche Investigaciones recientes han demostrado que las enzimas isoformes CYP4 podrían ayudar a reducir la sobre acumulación de ácido fitánico in vivo.
La plasmaféresis es otra intervención médica utilizada para tratar a los pacientes. Esto implica el filtrado de la sangre para asegurar que no haya acumulación de ácido fitánico.
Fuentes biológicas de ácido fitánico
En los rumiantes, la fermentación intestinal de los materiales vegetales consumidos libera fitol, un componente de la clorofila, que luego se convierte en ácido fitánico y se almacena en grasas.
Aunque los humanos no pueden derivar cantidades significativas de ácido fitánico del consumo de clorofila presente en los materiales vegetales, se ha propuesto que los grandes simios (bonobos, chimpancés, gorilas y orangutanes) pueden derivar cantidades significativas de ácido fitánico de la fermentación del intestino posterior de los materiales vegetales.

Dermatologa. Especialista en enfermedades de la piel, pelo y uñas.