El síndrome de Zinsser-Cole-Engman también conocido como disqueratosis congénita (DKC), es un raro trastorno congénito progresivo con un fenotipo altamente variable La entidad fue definida clásicamente por la tríada de pigmentación anormal de la piel, distrofia de uñas y leucoplasia de la mucosa oral, pero estos componentes no siempre ocurren.
La DKC se caracteriza por telómeros cortos. Algunas de las manifestaciones se asemejan al envejecimiento prematuro (similar a la progeria). Inicialmente, la enfermedad afecta principalmente a la piel, pero una consecuencia importante es la insuficiencia progresiva de la médula ósea, que se produce en más del 80%, causando una mortalidad prematura.
Características
La DKC se puede caracterizar por pigmentación cutánea, encanecimiento prematuro, distrofia de las uñas, leucoplasia de la mucosa oral, lagrimeo continuo debido a atresia de los conductos lagrimales, a menudo trombocitopenia, anemia, atrofia testicular en los portadores masculinos y predisposición al cáncer.
Muchos de estos síntomas son característicos de la geriatría, y aquellos que portan las formas más graves de la enfermedad a menudo han acortado significativamente la duración de la vida.
Características clínicas
Edad: Las características mucocutáneas del síndrome de Zinsser-Cole-Engman se desarrollan típicamente entre los 5 y 15 años de edad, la edad media de inicio de la citopenia periférica es de 10 años.
Sexo: La proporción de hombres y mujeres es de aproximadamente.
Físico: La tríada de hiperpigmentación reticulada de la piel, distrofia de uñas y leucoplasia caracteriza a la DKC. El síndrome es clínicamente heterogéneo; además de las características mucocutáneas diagnósticas y la insuficiencia de la médula ósea, los individuos afectados pueden tener una variedad de otras características clínicas.
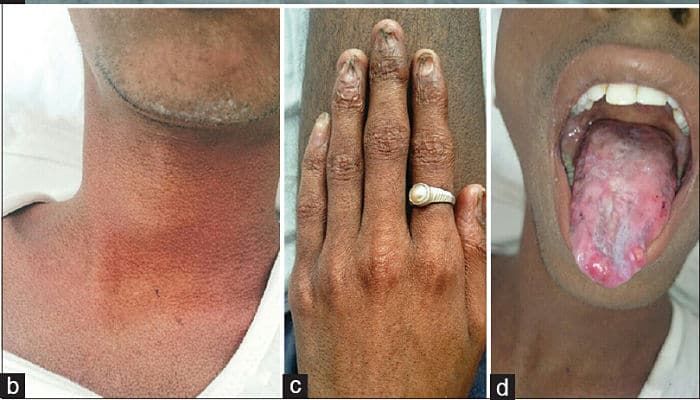
Hallazgos cutáneos
El hallazgo primario es la pigmentación anormal de la piel, con máculas y parches hiperpigmentadas o hipopigmentadas de color gris a gris en un patrón moteado o reticulado.
- La pigmentación reticulada ocurre en aproximadamente el 90% de los pacientes.
- Los cambios poiquilodérmicos con atrofia y telangiectasia son comunes.
- La presentación cutánea puede parecerse clínica e histológicamente a la enfermedad de injerto contra huésped.
- La distribución típica involucra las áreas expuestas al sol, incluyendo la parte superior del tronco, cuello y cara.
Otros hallazgos cutáneos pueden incluir alopecia del cuero cabelludo, cejas y pestañas; encanecimiento prematuro del cabello; hiperhidrosis; hiperqueratosis de las palmas de las manos y las plantas de los pies; y adermatoglifos (pérdida de crestas dérmicas en los dedos de las manos y de los pies).
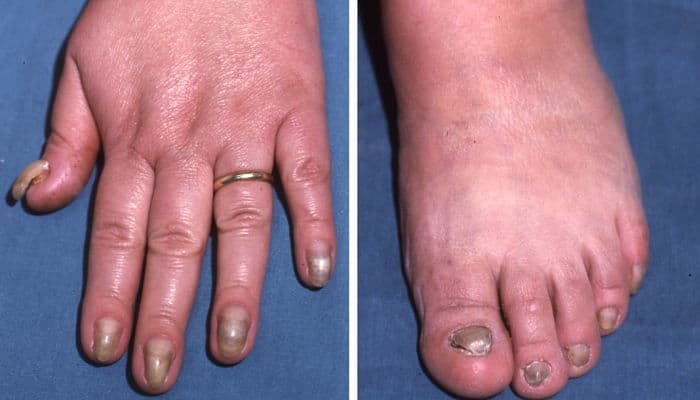
Descubrimiento de uñas
La distrofia de uña se observa en aproximadamente el 90% de los pacientes, con compromiso de la uña a menudo previo al compromiso de la uña del pie. La distrofia progresiva de las uñas comienza con la formación de surcos y la separación longitudinal. Atrofia progresiva, adelgazamiento, pterigión y distorsión eventual en uñas pequeñas, rudimentarias o ausentes.
Hallazgos de la mucosa
La leucoplasia mucosa ocurre en aproximadamente el 80% de los pacientes y típicamente involucra la mucosa bucal, la lengua y la orofaringe. La leucoplasia puede volverse verrugosa y se puede presentar ulceración. Los pacientes también pueden tener una mayor prevalencia y gravedad de la enfermedad periodontal.
Otros sitios de la mucosa pueden estar comprometidos (por ejemplo, esófago, meato uretral, glande del pene, conducto lagrimal, conjuntiva, vagina, ano). La constricción y la estenosis pueden ocurrir en estos sitios, con el desarrollo subsecuente de disfagia, disuria, fimosis y epífora.
Insuficiencia de la médula ósea
- Aproximadamente el 90% tiene citopenia periférica de uno o más linajes.
- En algunos casos, esta es la presentación inicial, con una edad media de inicio de 10 años.
- La insuficiencia de la médula ósea es una de las principales causas de muerte, con aproximadamente el 70% de las muertes relacionadas con el sangrado y las infecciones oportunistas como resultado de la insuficiencia de la médula ósea.
Complicaciones pulmonares
Aproximadamente el 20% de los individuos con síndrome de Zinsser-Cole-Engman desarrollan complicaciones pulmonares, incluyendo fibrosis pulmonar y anormalidades de la vasculatura pulmonar.
La recomendación es que los pacientes eviten tomar medicamentos con toxicidad pulmonar (por ejemplo, busulfan) y que tengan sus pulmones protegidos de la radiación durante el trasplante de médula ósea (BMT).
Aumento del riesgo de malignidad
Las pacientes tienen una mayor prevalencia de neoplasias malignas de la mucosa, particularmente carcinoma de células escamosas de la boca, nasofaringe, esófago, recto, vagina o cuello uterino.
Éstos a menudo ocurren dentro de los sitios de leucoplasia. La prevalencia del carcinoma escamocelular de la piel también aumenta. Otras neoplasias malignas reportadas incluyen linfoma de Hodgkin, adenocarcinoma del tracto gastrointestinal y carcinoma bronquial y laríngeo. La malignidad tiende a desarrollarse en la tercera década de la vida.
Hallazgos del sistema neurológico
- Los pacientes pueden tener dificultades de aprendizaje y retraso mental.
Hallazgos del sistema oftálmico
- Se ha informado que la DKC está asociada con conjuntivitis, blefaritidos y pterigión.
- La estenosis del conducto lagrimal que causa epífora (es decir, lagrimeo excesivo) ocurre en aproximadamente el 80% de los pacientes.
Hallazgos del sistema esquelético
- Los pacientes pueden tener hipoplasia mandibular, osteoporosis, necrosis vascular y escoliosis.
Hallazgos del sistema gastrointestinal
- Estos pueden incluir telarañas esofágicas, hepatoesplenomegalia, enteropatía y cirrosis.
Hallazgos del sistema genitourinario
- Se reportan testículos hipospásicos, hipospadias y estenosis ureteral.
Mujeres portadoras

Las mujeres portadoras de síndrome de Zinsser-Cole-Engman pueden tener características clínicas sutiles. Un estudio mostró que 3 de 20 mujeres portadoras tenían características clínicas que incluían una sola uña distrófica, un parche de hipopigmentación, o leucoplasia leve.
Fisiopatología
El Síndrome de Zinsser-Cole-Engman o disqueratosis congénita es un trastorno de mantenimiento deficiente de los telómeros debido principalmente a una serie de mutaciones genéticas que dan lugar a una función anormal del ribosoma, denominada ribosomopatía.
Específicamente, la enfermedad está relacionada con una o más mutaciones que afectan directa o indirectamente al componente de ARN de la telomerasa vertebrada (TERC).
La telomerasa es una transcriptasa inversa que mantiene una secuencia de repetición específica de ADN, el telómero, durante el desarrollo.
Los telómeros son colocados por la telomerasa en ambos extremos de los cromosomas lineales como una forma de proteger el ADN lineal de las formas generales de daño químico y de corregir el acortamiento del extremo cromosómico que ocurre durante la replicación normal del ADN.
Este acortamiento del extremo es el resultado de que las polimerasas de ADN eucariotas no tienen ningún mecanismo para sintetizar los nucleótidos finales presentes en el extremo de la “hebra retardada” del ADN de doble cadena.
La ADN polimerasa sólo puede sintetizar ADN nuevo a partir de una vieja cadena de ADN en la dirección 5’→’. Dado que el ADN tiene dos hebras que son complementarias, una debe ser 5’→’ mientras que la otra es 3’→’. Esta incapacidad de sintetizar en la direccionalidad 3’→’ se compensa con el uso de fragmentos de Okazaki, piezas cortas de ADN que se sintetizan 5’→’ del 3’→’ a medida que se mueve la horquilla de replicación.
Como la ADN polimerasa requiere primeros de ARN para la unión del ADN con el fin de comenzar la replicación, cada fragmento de Okazaki es precedido por un primer de ARN en la cadena que se está sintetizando.
Cuando se alcanza el final del cromosoma, el primer final del ARN se coloca sobre esta región nucleótida, y se elimina inevitablemente. Desafortunadamente una vez que la imprimación es removida, la DNA polimerasa es incapaz de sintetizar las bases restantes.
Se ha demostrado que los que sufren de DKC tienen una reducción en los niveles de TERC que afecta invariablemente la función normal de la telomerasa que mantiene estos telómeros.
Con niveles de TERC bajos, el mantenimiento de los telómeros durante el desarrollo sufre en consecuencia. En los humanos, la telomerasa es inactiva en la mayoría de los tipos de células después del desarrollo temprano (excepto en casos extremos como el cáncer)
Por lo tanto, si la telomerasa no es capaz de afectar eficientemente el ADN al principio de la vida, la inestabilidad cromosómica se convierte en una posibilidad grave en los individuos mucho antes de lo esperado.
Un estudio muestra que los defectos proliferativos en los queratinocitos cutáneos de CD se corrigen mediante la expresión de la telomerasa transcriptasa inversa, TERT, o mediante la activación de la telomerasa endógena a través de la expresión del virus del papiloma E6/E7 del componente de ARN de la telomerasa, TERC.
Genética

De los componentes del componente de ARN de la telomerasa (TERC), uno de importancia clave es la caja H/ACA dominio. Este dominio H/ACA es responsable de la maduración y estabilidad de la TERC y por lo tanto de la telomerasa en su conjunto. La ribonucleoproteína de mamíferos H/ACA ribonucleoproteína contiene cuatro subunidades de proteínas:
- Disquerina, Gar1, Nop10 y Nhp2.
- Se ha demostrado que las mutaciones en Nop10, Nhp2 y dyskerin1 conducen a síntomas similares a los de la DKC.
Ligado al cromosoma X
La forma mejor caracterizada de disqueratosis congénita es el resultado de una o más mutaciones en el brazo largo del cromosoma X en el gen DKC1. Esto da lugar a la forma recesiva ligada al cromosoma X de la enfermedad en la que la principal proteína afectada es la disquerina.
De las cinco mutaciones descritas por Heiss y sus colegas en Nature Genéticas, cuatro eran polimorfismos de nucleótidos simples que resultaron en el cambio de aminoácidos altamente conservados. Un caso fue una eliminación dentro del marco que resultó en la pérdida de un residuo de leucina, también conservado en mamíferos.
En tres de los casos, los aminoácidos específicos afectados (fenilalanina, prolina, glicina) se encuentran en el mismo lugar en humanos que en la levadura (S. Cerevisiae) y la rata parda (R. Norvegicus), lo que establece la conservación de la secuencia y la importancia de la disquerina dentro de los eucariontes.
La naturaleza relevante de la disquerina en la mayoría de las especies es catalizar la pseudouridilación post-transcripcional de uridinas específicas encontradas en ARNs no codificadores, como el ARN ribosomal (rRNA). Cbf5, el análogo de la levadura de la disquerina humana, es de hecho conocido por estar asociado con el procesamiento y maduración del rRNA.
En humanos este papel puede ser atribuido a la disquerina[8] Por lo tanto, la forma ligada al cromosoma X de esta enfermedad puede resultar en problemas específicos relacionados con el rRNA disfuncional y quizás un fenotipo más grave.
Dentro de los vertebrados, a diferencia de los eucariontes unicelulares, la disquerina es un componente clave del componente de ARN de la telomerasa (TERC) en forma de motivo H/ACA. Esta variedad ligada al cromosoma X, al igual que las mutaciones Nop10 y Nhp2, demuestra telómeros acortados como resultado de menores concentraciones de TERC.
Autosómico dominante de el síndrome de Zinsser-Cole-Engman
3 genes: TERC, TERT, TINF2
La evidencia que apoya la importancia del dominio H/ACA en la telomerasa humana es abundante. Al menos un estudio ha demostrado que estas mutaciones afectan la actividad de la telomerasa al afectar negativamente el ensamblaje previo al PNR y la maduración del ARN de la telomerasa humana.
No obstante, las mutaciones que afectan directamente a los componentes del ARN de la telomerasa presumiblemente existirían y también deberían causar envejecimiento prematuro o síntomas similares a los de la DKC. De hecho, tres familias con mutaciones en el gen humano TERC han sido estudiadas con resultados intrigantes.
En dos de estas familias, dos polimorfismos de nucleótidos únicos específicos de la familia estaban presentes, mientras que en la otra persistió una deleción a gran escala (821 pares de bases de ADN) en el cromosoma 3 que incluye 74 bases que codifican una sección del dominio H/ACA.
Estas tres mutaciones diferentes resultan en una forma leve de disqueratosis congénita que sigue de manera única un patrón autosómico dominante de herencia. El encanecimiento prematuro, la pérdida dental temprana, la predisposición al cáncer de piel, así como el acortamiento de la longitud de los telómeros siguen siendo característicos de esta enfermedad.
Autosómico recesivo
6 genes
El verdadero fenotipo de los individuos con DKC puede depender de qué proteína ha incurrido en una mutación. Una mutación autosómica recesiva documentada en una familia portadora de DKC se ha encontrado en Nop10, específicamente, la mutación es un cambio de base de citosina a timina en una región altamente conservada de la secuencia Nop10.
Esta mutación, en el cromosoma 15, resulta en un cambio de aminoácidos de arginina a triptófano, los individuos homocigóticos recesivos muestran los síntomas de la disqueratosis congénita en su totalidad. En comparación con los individuos normales de la misma edad, los que sufren de DKC tienen telómeros de una longitud mucho más corta.
Además, los heterocigotos, aquellos que tienen un alelo normal y una codificación para la enfermedad, también muestran telómeros relativamente acortados. Se determinó que la causa de esto era una reducción en los niveles de TERC en aquellos con la mutación Nop10.
Con la reducción de los niveles de TERC, se supondría que el mantenimiento de los telómeros, especialmente en desarrollo, se vería afectado en consecuencia, esto llevaría al acortamiento de los telómeros descrito. Las mutaciones de Nhp2 son similares en caracterización a Nop10. Estas mutaciones son también autosómicas recesivas con tres polimorfismos específicos de un solo nucleótido reconocidos que resultan en disqueratosis congénita.
También como Nop10, los individuos con estas mutaciones Nhp2 tienen una reducción en la cantidad de componente de ARN de la telomerasa (TERC) presente en la célula. Una vez más se puede suponer que una reducción en la TERC resulta en un mantenimiento aberrante de los telómeros y, por lo tanto, en un acortamiento de los telómeros. Los homocigóticos recesivos para mutaciones en Nhp2 muestran telómeros más cortos cuando se comparan con individuos normales de la misma edad.
Predisposición al cáncer
La susceptibilidad al cáncer parece contraintuitiva porque en muchos cánceres conocidos la reactivación de la telomerasa es en realidad un paso necesario para que la malignidad evolucione (ver telómero). En una enfermedad en la que la telomerasa está afectada, no parece que el cáncer sea una complicación.
Los autores señalan la naturaleza paradójica de la predisposición al cáncer en individuos que parecen carecer de uno de los componentes necesarios para que se forme el cáncer. Se piensa que sin telomerasa funcional, es probable que los cromosomas se unan entre sí en sus extremos a través de la vía de unión final no homóloga. Si esto resulta ser una ocurrencia lo suficientemente común, es posible que se presente malignidad incluso sin la presencia de telomerasa.
Conclusión
La disqueratosis congénita (DKC, por sus siglas en inglés) es un trastorno multisistémico grave asociado con mortalidad prematura que generalmente se debe a la insuficiencia/inmunodeficiencia de la médula ósea (BM, por sus siglas en inglés).
El dentista debe ser capaz de reconocer esta condición fatal en sus primeras etapas y aconsejar investigaciones hematológicas apropiadas. Los dentistas pueden ser los primeros en ver y diagnosticar la DKC y tienen un papel importante en el monitoreo de los cambios malignos orales en la mucosa.
También podrías leer: Síndrome ABCD: Clasificación, Síntomas, Causas, Revisión, Tratamiento

Dermatologa. Especialista en enfermedades de la piel, pelo y uñas.